Obecný přehled EU FMD
Se směrnicí o padělaných léčivech (FMD) zveřejněnou v červenci 2011, Evropská léková agentura (EMA) nastavit počátek standardů sledovatelnosti zemí Evropské unie.
The Směrnice o padělaných léčivech (směrnice 2011/62/EU) zavádí harmonizovaná evropská opatření pro boj proti padělání léků a zajištění bezpečnosti léků a přísné kontroly léků v obchodním oběhu. Mezi opatření patří:
- Povinné bezpečnostní prvky – jedinečný identifikátor a zařízení proti neoprávněné manipulaci – na vnějším obalu léků
- Společné logo pro celou EU k identifikaci legálních online lékáren
- Přísnější pravidla pro dovoz účinných farmaceutických látek
- Posílení požadavků na vedení záznamů pro velkoobchodní distributory
Od roku 2019 musí všechny farmaceutické produkty plně splňovat povinnosti slintavky a kulhavky. Do roku 2025 musí země EU se samostatným systémem, jako je Řecko a Itálie, plně dodržovat nařízení o sledování a sledování léčiv.
Požadavky na serializaci
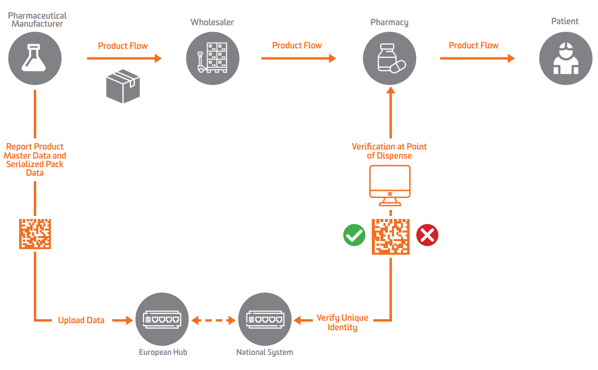
Podle EU FMD se serializace musí v Evropě objevit na úrovni sekundární nebo prodejné jednotky. Aby bylo možné ověření umožnit, musí výrobci nejprve serializovat produkt a odeslat tato serializovaná data do centrálního úložiště, které proti nim může provádět dotazy.
Pro umožnění serializace, ověřování a podávání zpráv úřadům vyžaduje EU FMD, aby výrobci označili balíčky čtyřmi datovými prvky, které musí být vytištěny ve formě čitelné pro člověka a zakódovány a uloženy v GS1 2D DataMatrix:
- Identifikátor produktu
- Sériové číslo
- Číslo šarže nebo šarže
- Datum vypršení platnosti
Použití pátého datového prvku – národního čísla úhrady – je dobrovolné a jen málo států v EU může požádat o uvedení stejného identifikátoru v jedinečném identifikátoru pro propojení úhrady léku v rámci programu socializované medicíny.
Výrobci léčiv a paralelní obchodníci musí hlásit data do centrálního EU Hub provozuje Evropská organizace pro ověřování léčiv (EMVO), který také zapojuje uživatele do Hubu. To přesune data do příslušných datových úložišť provozovaných příslušnými národními organizacemi pro ověřování léčiv (NMVO), které jsou odpovědné za registraci koncových uživatelů a za provoz národních systémů.
Požadavky na podávání zpráv
Podle EU FMD je držitel rozhodnutí o registraci (MAH) povinen předložit kmenová data produktu a serializovaná data balení produktu.
Kmenová data zahrnují:
- Kódy produktů
- Formulář
- Síla
- Dávky v balení
- Typ balení
- Cílový trh (trhy) pro distribuci
- Budoucí a jakékoli aktualizace kmenových dat produktu
Serializovaná data produktového balíčku zahrnují:
- Kódy produktů
- Číslo šarže/šarže
- Datum vypršení platnosti
- Sériová čísla
- Jakékoli aktualizace serializovaných dat produktových balíčků