Ιατρικές συσκευές
Η ιχνηλασιμότητα διαδραματίζει βασικό ρόλο στην παρακολούθηση, την επίβλεψη και τη βελτίωση της συνολικής ποιότητας των ιατροτεχνολογικών προϊόντων και των συσκευών in vitro.
Κανονισμοί ΕΕ
Το 2017 το Κανονισμός για ιατροτεχνολογικά προϊόντα (MDR) δημοσιεύτηκε και σηματοδότησε την έναρξη ενός τετραετούς ταξιδιού μετάβασης από το MDD στο AIMDD. Από τις 26 Μαΐου 2021, το MDR θα ισχύει πλήρως. Η MDR επηρεάζει τις βιομηχανίες κατασκευής, διανομής ή προμήθειας ιατροτεχνολογικών προϊόντων.
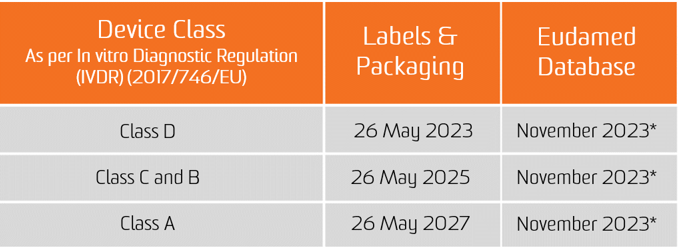
ο Διαγνωστικός κανονισμός in vitro (IVDR) τέθηκε σε ισχύ στις 25 Μαΐου 2017. Από τις 26 Μαΐου 2022 οι νέες συσκευές θα πρέπει να πληρούν τις απαιτήσεις του IVDR προκειμένου να διατεθούν στην ευρωπαϊκή αγορά. Προϊόντα που έχουν ήδη πιστοποιηθεί από Κοινοποιημένο Οργανισμό μπορούν να διατεθούν στην αγορά έως τις 25 Μαΐου 2024 υπό ορισμένες προϋποθέσεις και εάν ο κατασκευαστής πληροί τις ειδικές προαπαιτούμενες απαιτήσεις που ορίζονται στο IVDR.
Τι είναι το UDI;
UDI (Μοναδικός Αναγνώριση συσκευής)
ο UDI είναι μια σειρά αριθμητικών ή αλφαριθμητικών χαρακτήρων που δημιουργούνται μέσω ενός παγκοσμίως αποδεκτού προτύπου αναγνώρισης και κωδικοποίησης συσκευής. Επιτρέπει τη σαφή αναγνώριση μιας συγκεκριμένης συσκευής στην αγορά. Το UDI αποτελείται από το UDI-DI και το UDI-PI.
 UDI-DI (Ισοδύναμο GS1 GTIN- Παγκόσμιος αριθμός αντικειμένου εμπορίου)
UDI-DI (Ισοδύναμο GS1 GTIN- Παγκόσμιος αριθμός αντικειμένου εμπορίου)
Το UDI-DI είναι ένας μοναδικός αριθμητικός ή αλφαριθμητικός κώδικας συγκεκριμένος για ένα μοντέλο της συσκευής και χρησιμοποιείται επίσης ως «κλειδί πρόσβασης» στις πληροφορίες που είναι αποθηκευμένες σε μια βάση δεδομένων UDI.
UDI- PI (GS1 Equivalent AI – Application Identifiers)
Το UDI-PI είναι ένας αριθμητικός ή αλφαριθμητικός κωδικός που προσδιορίζει τη μονάδα παραγωγής της συσκευής.
Οι διαφορετικοί τύποι UDI-PI περιλαμβάνουν σειριακό αριθμό, αριθμό παρτίδας, αναγνώριση λογισμικού και ημερομηνία κατασκευής ή λήξης ή και τους δύο τύπους δεδομένων.
Υλοποίηση UDI
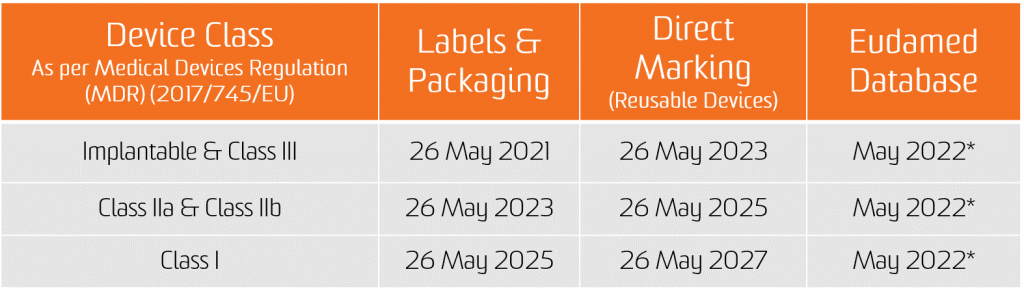
Η υποχρέωση για την ανάθεση UDI ισχύει από την ημερομηνία εφαρμογής των δύο νέων Κανονισμών, ήτοι 26 Μαΐου 2021 για ιατροτεχνολογικά προϊόντα και 26 Μαΐου 2022 για In Vitro διαγνωστικά ιατροτεχνολογικά προϊόντα.
Η υποχρέωση υποβολής δεδομένων UDI στη βάση δεδομένων EUDAMED ισχύει από τις 26 Νοεμβρίου 2022 για ιατροτεχνολογικά προϊόντα και από τις 26 Νοεμβρίου 2023 για τα in vitro διαγνωστικά ιατροτεχνολογικά προϊόντα (υπό την προϋπόθεση ότι η EUDAMED είναι πλήρως λειτουργική πριν από την ημερομηνία εφαρμογής του αντίστοιχου κανονισμού· διαφορετικά, αυτή η υποχρέωση ισχύει 24 μήνες μετά την πλήρη λειτουργία του EUDAMED).
Ωστόσο, οι κατασκευαστές θα είναι σε θέση να συμμορφώνονται εθελοντικά με τις υποχρεώσεις καταχώρισης από τις 26 Μαΐου 2021 για τα ιατροτεχνολογικά προϊόντα και από τις 26 Μαΐου 2022 για τα διαγνωστικά ιατροτεχνολογικά προϊόντα In Vitro.
Σημειώνεται ότι, υπό την προϋπόθεση ότι το Eudamed είναι πλήρως λειτουργικό, ανά πάσα στιγμή μετά τις 26 Μαΐου 2021 για τα ιατροτεχνολογικά προϊόντα και τις 26 Μαΐου 2022 για τα διαγνωστικά ιατροτεχνολογικά προϊόντα In Vitro, η πλήρης καταχώριση των συσκευών (άρθρο 29 MDR και άρθρο 26 IVDR) παραμένει προϋπόθεση για την πιθανή καταγραφή του σχετικού σοβαρού περιστατικού τους στο Eudamed.
*Το έγγραφο CAMD στις 11 Νοεμβρίου 2020 αναφέρει ότι η Ευρωπαϊκή Επιτροπή ανακοινώνει την καθυστέρηση της κυκλοφορίας του Eudamed συνολικά μέχρι τον Μάιο του 2022 για νομικούς λόγους. Η νέα ημερομηνία κυκλοφορίας του Eudamed συμπίπτει με την ημερομηνία εφαρμογής του Διαγνωστικού Κανονισμού In Vitro, ο οποίος θα τεθεί σε ισχύ στις 26 Μαΐου 2022.
Τι είναι το EUDAMED;
Η νέα κανονιστική ευρωπαϊκή βάση δεδομένων για τα ιατροτεχνολογικά προϊόντα
Το Eudamed είναι μια ασφαλής διαδικτυακή πύλη που λειτουργεί ως κεντρικός χώρος αποθήκευσης για την ανταλλαγή πληροφοριών μεταξύ των εθνικών αρμόδιων αρχών και της Επιτροπής σύμφωνα με τους κανονισμούς MDR & IVDR.
Ο κύριος ρόλος του Eudamed περιλαμβάνει τις ακόλουθες αρμοδιότητες:
![]() Εγγραφή Eudamed
Εγγραφή Eudamed
![]() Εγγραφή UDI/Συσκευών
Εγγραφή UDI/Συσκευών
![]() Λήψη & Προβολή Πιστοποιητικών από Κοινοποιημένους Φορείς
Λήψη & Προβολή Πιστοποιητικών από Κοινοποιημένους Φορείς
![]() Παρέχετε δεδομένα κλινικών ερευνών και μελετών απόδοσης
Παρέχετε δεδομένα κλινικών ερευνών και μελετών απόδοσης
![]() Αναφέρετε επαγρύπνηση και επιτήρηση μετά τη διάθεση στην αγορά
Αναφέρετε επαγρύπνηση και επιτήρηση μετά τη διάθεση στην αγορά
![]() Δραστηριότητες εποπτείας της αγοράς
Δραστηριότητες εποπτείας της αγοράς
Παρακολουθήστε τα διαδικτυακά σεμινάρια all-in-one για τους κανονισμούς ιατρικών συσκευών
[Webcast]: Ικανοποίηση της απαίτησης UDI για ιατρικές συσκευές από την GS1 και το SoftGroup

Ζητήστε τη διαδικτυακή μετάδοση