Présentation générale de l'EU FMD
Avec la directive sur les médicaments falsifiés (FMD) publiée en juillet 2011, le Agence européenne des médicaments (EMA) fixé le début des normes de traçabilité des pays de l'Union européenne.
Le Directive sur les médicaments falsifiés (directive 2011/62/UE) introduit des mesures européennes harmonisées pour lutter contre les falsifications de médicaments et garantir que les médicaments sont sûrs et que les médicaments en circulation sont strictement contrôlés. Les mesures comprennent :
- Dispositifs de sécurité obligatoires - un identifiant unique et un dispositif anti-effraction - sur l'emballage extérieur des médicaments
- Un logo commun à l'échelle de l'UE pour identifier les pharmacies en ligne légales
- Des règles plus strictes sur l'importation d'ingrédients pharmaceutiques actifs
- Exigences renforcées en matière de tenue de registres pour les distributeurs en gros
Depuis 2019, tous les produits pharmaceutiques doivent satisfaire pleinement aux obligations de la FMD. Jusqu'en 2025, les pays de l'UE dotés d'un système distinct, tels que la Grèce et l'Italie, doivent se conformer pleinement au règlement sur le suivi et la traçabilité des produits pharmaceutiques.
Exigences de sérialisation
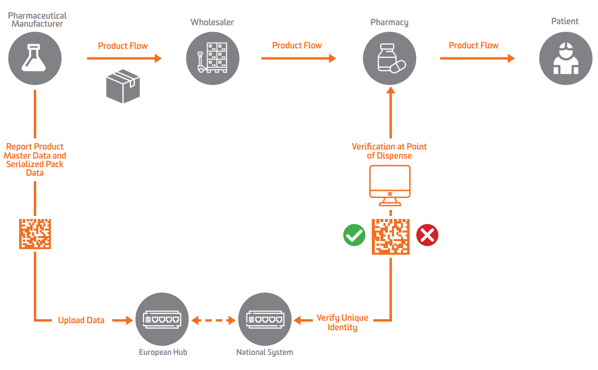
Selon l'EU FMD, la sérialisation doit apparaître au niveau de l'unité secondaire ou vendable en Europe. Pour activer la vérification, les fabricants doivent d'abord sérialiser le produit et envoyer ces données sérialisées à un référentiel central qui peut effectuer des requêtes sur celui-ci.
Pour permettre la sérialisation, la vérification et la déclaration aux autorités, l'EU FMD exige que les fabricants marquent les colis avec quatre éléments de données qui doivent être imprimés sous une forme lisible par l'homme, puis codés et stockés dans un DataMatrix GS1 2D :
- étiquette d'un produit
- Numéro de série
- Numéro de lot ou lot
- Date d'expiration
L’utilisation du cinquième élément de données – le numéro national de remboursement – est facultative, et peu d’États de l’UE peuvent demander à l’inclure dans un identifiant unique pour lier le remboursement d’un médicament dans le cadre d’un programme de médecine socialisée.
Les fabricants de produits pharmaceutiques et les commerçants parallèles doivent transmettre les données à un hub central de l'UE dirigé par le Organisation européenne de vérification des médicaments (EMVO), qui intègre également les utilisateurs dans le Hub. Cela permettra de transférer les données vers les référentiels de données appropriés gérés par les organisations nationales de vérification des médicaments (NMVO) correspondantes, qui sont responsables de l'intégration des utilisateurs finaux et du fonctionnement des systèmes nationaux.
Exigences en matière de rapports
Dans le cadre de l'EU FMD, le titulaire de l'autorisation de mise sur le marché (MAH) est tenu de soumettre les données de base du produit et les données sérialisées de l'emballage du produit.
Les données de base comprennent :
- Codes produit
- Former
- Force
- Doses par paquet
- Type d'emballage
- Marché(s) cible(s) pour la distribution
- Futures et toutes mises à jour des données de base du produit
Les données sérialisées des packs de produits incluent :
- Codes produit
- Numéro de lot
- Date d'expiration
- Numéros de série
- Toute mise à jour des données sérialisées des packs de produits