Dispositivi medici
La tracciabilità ha un ruolo chiave nel monitoraggio, nella supervisione e nel miglioramento della qualità complessiva dei dispositivi medici e dei dispositivi in vitro.
Regolamento UE
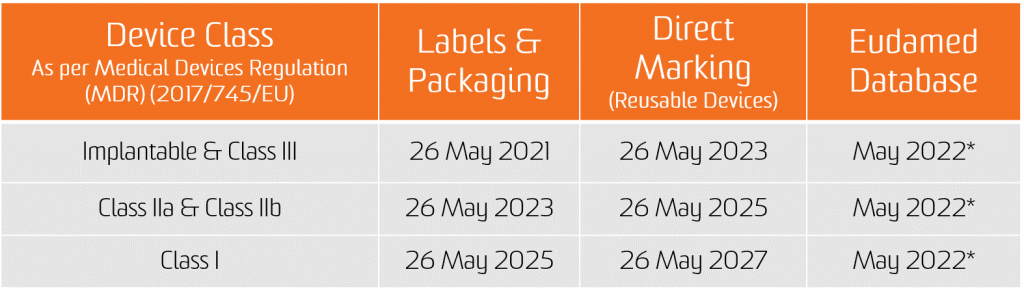
Nel 2017 il Regolamento sui dispositivi medici (MDR) è stato pubblicato e ha segnato l'inizio di un viaggio di quattro anni di transizione da MDD ad AIMDD. Dal 26 maggio 2021, l'MDR sarà pienamente applicabile. L'MDR interessa le industrie di produzione, distribuzione o approvvigionamento di dispositivi medici.
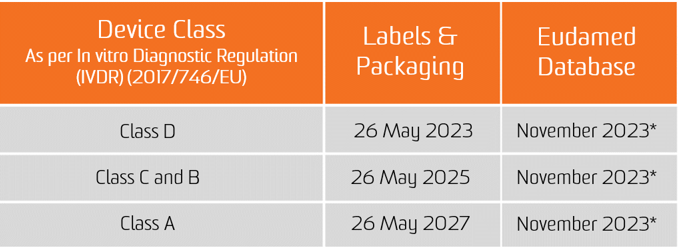
IL Regolamento diagnostico in vitro (IVDR) è entrato in vigore il 25 maggio 2017. Dal 26 maggio 2022 i nuovi dispositivi dovranno soddisfare i requisiti dell'IVDR per poter essere immessi nel mercato europeo. I prodotti già certificati da un Organismo Notificato possono essere immessi sul mercato fino al 25 maggio 2024 a determinate condizioni e se il produttore soddisfa i requisiti prerequisiti specifici previsti dall'IVDR.
Che cos'è l'UDI?
UDI (Unico Identificazione del dispositivo)
IL UDI è una serie di caratteri numerici o alfanumerici creati attraverso uno standard di identificazione e codifica del dispositivo accettato a livello globale. Consente l'identificazione univoca di uno specifico dispositivo sul mercato. L'UDI è composto dall'UDI-DI e dall'UDI-PI.
 UDI-DI (GTIN equivalente a GS1 - Numero articolo commerciale globale)
UDI-DI (GTIN equivalente a GS1 - Numero articolo commerciale globale)
L'UDI-DI è un codice numerico o alfanumerico univoco specifico per un modello del dispositivo e utilizzato anche come "chiave di accesso" alle informazioni memorizzate in una banca dati UDI.
UDI-PI (GS1 Equivalent AI – Identificatori dell'applicazione)
L'UDI-PI è un codice numerico o alfanumerico che identifica l'unità di produzione del dispositivo.
I diversi tipi di UDI-PI includono un numero di serie, un numero di lotto, l'identificazione del software e la data di produzione o di scadenza o entrambi i tipi di dati.
Implementazione dell'UDI
L'obbligo di assegnazione dell'UDI decorre dalla data di applicazione dei due nuovi Regolamenti, ovvero il 26 maggio 2021 per i dispositivi medici e il 26 maggio 2022 per i dispositivi medico-diagnostici in vitro.
L'obbligo di presentazione dei dati UDI nella banca dati EUDAMED si applica dal 26 novembre 2022 per i dispositivi medici e dal 26 novembre 2023 per i dispositivi medico-diagnostici in vitro (a condizione che EUDAMED sia pienamente funzionante prima della data di applicazione del rispettivo regolamento; in caso contrario, tale obbligo si applica 24 mesi dopo che EUDAMED è diventato pienamente operativo).
Tuttavia, i produttori potranno adempiere volontariamente agli obblighi di registrazione a partire dal 26 maggio 2021 per i dispositivi medici e dal 26 maggio 2022 per i dispositivi medico-diagnostici in vitro.
Si precisa che, a condizione che Eudamed sia pienamente funzionante, in qualsiasi momento successivo al 26 maggio 2021 per i dispositivi medici e al 26 maggio 2022 per i dispositivi medico-diagnostici in vitro, la piena registrazione dei dispositivi (art. 29 MDR e art. 26 IVDR) rimane un prerequisito per l'eventuale registrazione del loro incidente grave rilevante in Eudamed.
* Il documento CAMD dell'11 novembre 2020 afferma che la Commissione europea sta annunciando il rinvio del lancio di Eudamed nel suo complesso fino a maggio 2022 per motivi legali. La nuova data di lancio di Eudamed coincide con la data di attuazione del regolamento sulla diagnostica in vitro, che entrerà in vigore il 26 maggio 2022.
Che cos'è EUDAMED?
La nuova banca dati normativa europea sui dispositivi medici
Eudamed è un portale Web sicuro che funge da archivio centrale per lo scambio di informazioni tra le autorità nazionali competenti e la Commissione in conformità con i regolamenti MDR e IVDR.
Il ruolo principale di Eudamed comprende le seguenti responsabilità:
![]() Iscrizione Eudamed
Iscrizione Eudamed
![]() Registrazione UDI/Dispositivi
Registrazione UDI/Dispositivi
![]() Ottenere e visualizzare certificati da organismi notificati
Ottenere e visualizzare certificati da organismi notificati
![]() Fornire indagini cliniche e dati sugli studi delle prestazioni
Fornire indagini cliniche e dati sugli studi delle prestazioni
![]() Segnalare la vigilanza e la sorveglianza post-vendita
Segnalare la vigilanza e la sorveglianza post-vendita
![]() Attività di sorveglianza del mercato
Attività di sorveglianza del mercato
Guarda i webinar all-in-one per i regolamenti sui dispositivi medici
[Webcast]: soddisfare i requisiti UDI per i dispositivi medici da parte di GS1 e SoftGroup

Richiedi il webcast