Medische apparaten
Traceerbaarheid speelt een sleutelrol bij de bewaking, supervisie en verbetering van de algehele kwaliteit van medische hulpmiddelen en in-vitroapparatuur.
EU-regelgeving
In 2017 de Verordening Medische Hulpmiddelen (MDR) werd gepubliceerd en markeerde het begin van een vier jaar durende transitie van MDD naar AIMDD. Vanaf 26 mei 2021 is de MDR volledig van toepassing. De MDR is van invloed op de industrieën van productie, distributie of aanschaf van medische hulpmiddelen.
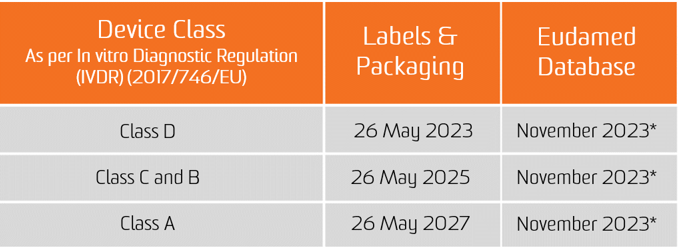
De In vitro Diagnostische Regeling (IVDR) is op 25 mei 2017 in werking getreden. Vanaf 26 mei 2022 moeten nieuwe apparaten voldoen aan de eisen van de IVDR om op de Europese markt te mogen worden gebracht. Producten die al zijn gecertificeerd door een aangemelde instantie mogen tot 25 mei 2024 op de markt worden gebracht onder bepaalde voorwaarden en als de fabrikant voldoet aan de specifieke randvoorwaarden die zijn opgesteld in de IVDR.
Wat is UDI?
UDI (Uniek apparaat identificatie)
De UDI is een reeks numerieke of alfanumerieke tekens die zijn gemaakt via een wereldwijd geaccepteerde apparaatidentificatie- en coderingsstandaard. Het maakt de ondubbelzinnige identificatie van een specifiek apparaat op de markt mogelijk. De UDI bestaat uit de UDI-DI en de UDI-PI.
 UDI-DI (GS1-equivalent GTIN- artikelnummer voor wereldwijde handel)
UDI-DI (GS1-equivalent GTIN- artikelnummer voor wereldwijde handel)
De UDI-DI is een unieke numerieke of alfanumerieke code die specifiek is voor een model van het apparaat en die ook wordt gebruikt als 'toegangssleutel' tot de informatie die is opgeslagen in een UDI-database.
UDI-PI (GS1-equivalente AI – Applicatie-ID's)
De UDI-PI is een numerieke of alfanumerieke code die de productie-eenheid van het hulpmiddel identificeert.
De verschillende soorten UDI-PI's omvatten een serienummer, partijnummer, software-identificatie en productie- of vervaldatum, of beide soorten gegevens.
UDI-implementatie
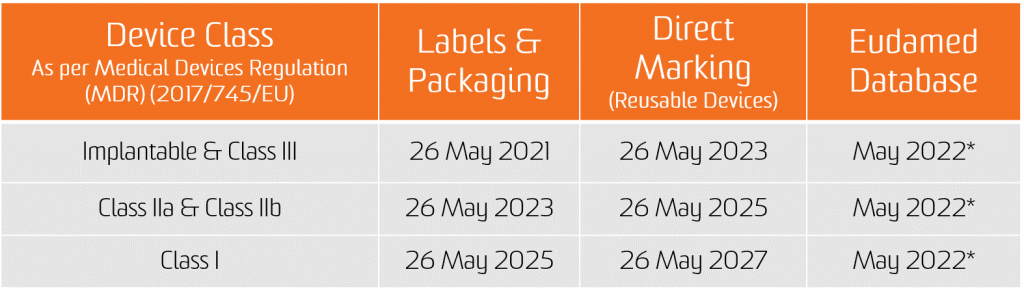
De verplichting voor de UDI-toekenning geldt vanaf de datum van toepassing van de twee nieuwe verordeningen, zijnde 26 mei 2021 voor medische hulpmiddelen en 26 mei 2022 voor medische hulpmiddelen voor in-vitrodiagnostiek.
De verplichting tot het indienen van UDI-gegevens in de EUDAMED-databank geldt vanaf 26 november 2022 voor medische hulpmiddelen en vanaf 26 november 2023 voor medische hulpmiddelen voor in-vitrodiagnostiek (op voorwaarde dat EUDAMED volledig functioneel is vóór de datum van toepassing van de betreffende verordening; anders is deze verplichting is van toepassing 24 maanden nadat EUDAMED volledig functioneel is geworden).
Fabrikanten zullen echter vanaf 26 mei 2021 voor medische hulpmiddelen en vanaf 26 mei 2022 voor medische hulpmiddelen voor in-vitrodiagnostiek vrijwillig kunnen voldoen aan de registratieverplichtingen.
Opgemerkt wordt dat, op voorwaarde dat Eudamed volledig functioneel is, op enig moment na 26 mei 2021 voor medische hulpmiddelen en 26 mei 2022 voor medische hulpmiddelen voor in-vitrodiagnostiek, de volledige registratie van hulpmiddelen (artikel 29 van de MDR en artikel 26 van de IVDR) voorwaarde blijft voor de eventuele registratie van hun relevante ernstige incident in Eudamed.
*CAMD-nummer op 11 november 2020-document stelt dat de Europese Commissie om juridische redenen de vertraging van de lancering van Eudamed als geheel tot mei 2022 aankondigt. De nieuwe lanceringsdatum voor Eudamed valt samen met de implementatiedatum voor de In Vitro Diagnostic Regulation, die op 26 mei 2022 van kracht wordt.
Wat is EUDAMED?
De nieuwe regelgevende Europese databank voor medische hulpmiddelen
Eudamed is een beveiligd webportaal dat fungeert als centrale opslagplaats voor informatie-uitwisseling tussen nationale bevoegde autoriteiten en de Commissie in overeenstemming met de MDR- en IVDR-voorschriften.
De belangrijkste rol van Eudamed omvat de volgende verantwoordelijkheden:
![]() Eudamed-registratie
Eudamed-registratie
![]() UDI/Apparaten registratie
UDI/Apparaten registratie
![]() Verkrijg en bekijk certificaten van aangemelde instanties
Verkrijg en bekijk certificaten van aangemelde instanties
![]() Verstrek gegevens over klinische onderzoeken en prestatiestudies
Verstrek gegevens over klinische onderzoeken en prestatiestudies
![]() Waakzaamheid en toezicht na het in de handel brengen melden
Waakzaamheid en toezicht na het in de handel brengen melden
![]() Markttoezichtactiviteiten
Markttoezichtactiviteiten
Bekijk de alles-in-één webinars voor Regelgeving Medische Hulpmiddelen
[Webcast]: Voldoen aan de UDI-vereiste voor medische hulpmiddelen door GS1 en SoftGroup

Vraag de webcast aan