Медицинское оборудование
Прослеживаемость играет ключевую роль в мониторинге, надзоре и улучшении общего качества медицинских устройств и устройств in-vitro.
Правила ЕС
В 2017 году Постановление о медицинских устройствах (MDR) был опубликован и ознаменовал начало четырехлетнего перехода от MDD к AIMDD. С 26 мая 2021 года MDR будет применяться в полной мере. MDR влияет на отрасли производства, распространения или закупки медицинских устройств.
The Регламент диагностики in vitro (IVDR) вступил в силу 25 мая 2017 г. С 26 мая 2022 г. новые устройства должны будут соответствовать требованиям IVDR, чтобы их можно было разместить на европейском рынке. Продукты, уже сертифицированные уполномоченным органом, могут быть размещены на рынке до 25 мая 2024 года при определенных условиях и в том случае, если производитель выполнит конкретные предварительные требования, изложенные в IVDR.
Что такое УДИ?
УДИ (Уникальный Идентификация устройства)
The УДИ представляет собой серию цифровых или буквенно-цифровых символов, которые создаются с помощью общепринятого стандарта идентификации и кодирования устройств. Это позволяет однозначно идентифицировать конкретное устройство на рынке. UDI состоит из UDI-DI и UDI-PI.
 УДИ-ДИ (эквивалентный номер GTIN GS1 — глобальный номер предмета торговли)
УДИ-ДИ (эквивалентный номер GTIN GS1 — глобальный номер предмета торговли)
UDI-DI — это уникальный числовой или буквенно-цифровой код, характерный для модели устройства, который также используется в качестве «ключа доступа» к информации, хранящейся в базе данных UDI.
УДИ-ПИ (Эквивалент ИИ GS1 — идентификаторы приложений)
UDI-PI — это числовой или буквенно-цифровой код, идентифицирующий единицу продукции устройства.
Различные типы UDI-PI включают серийный номер, номер партии, идентификацию программного обеспечения и дату изготовления или истечения срока действия, или оба типа данных.
Реализация UDI
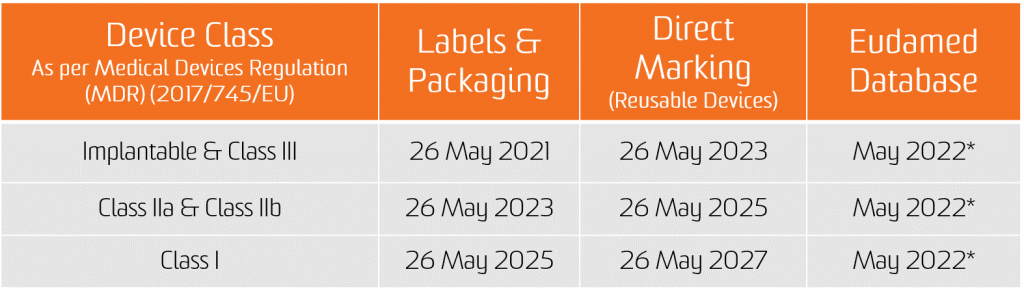
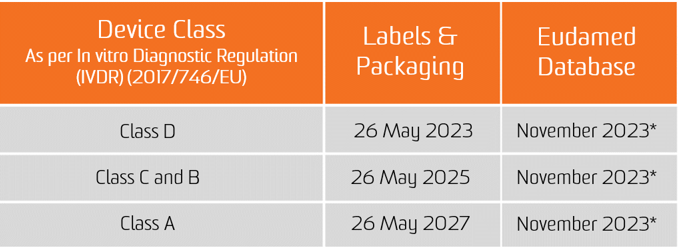
Обязательство по присвоению UDI применяется с даты применения двух новых Регламентов, то есть с 26 мая 2021 года для медицинских изделий и с 26 мая 2022 года для медицинских изделий для диагностики in vitro.
Обязательство по предоставлению данных UDI в базу данных EUDAMED применяется с 26 ноября 2022 г. для медицинских изделий и с 26 ноября 2023 г. для медицинских изделий для диагностики in vitro (при условии, что EUDAMED полностью функционирует до даты применения соответствующего Регламента; в противном случае это обязательство применяется через 24 месяца после того, как EUDAMED стал полностью функциональным).
Однако с 26 мая 2021 г. для медицинских изделий и с 26 мая 2022 г. для медицинских изделий для диагностики in vitro производители смогут добровольно выполнять обязательства по регистрации.
Следует отметить, что при условии полной работоспособности Eudamed в любое время после 26 мая 2021 г. для медицинских изделий и 26 мая 2022 г. для медицинских изделий для диагностики in vitro полная регистрация изделий (статья 29 MDR и статья 26 IVDR) остается предварительным условием для возможной регистрации их соответствующего серьезного инцидента в Eudamed.
* В выпуске CAMD от 11 ноября 2020 г. говорится, что Европейская комиссия объявляет об отсрочке запуска Eudamed в целом до мая 2022 г. по юридическим причинам. Новая дата запуска Eudamed совпадает с датой вступления в силу Регламента диагностики in vitro, который вступит в силу 26 мая 2022 года.
Что такое ЭУДАМЕД?
Новая нормативная европейская база данных по медицинским устройствам
Eudamed — это защищенный веб-портал, действующий в качестве центрального хранилища для обмена информацией между национальными компетентными органами и Комиссией в соответствии с положениями MDR и IVDR.
Основная роль Eudamed включает в себя следующие обязанности:
![]() Регистрация Евдамед
Регистрация Евдамед
![]() UDI/регистрация устройств
UDI/регистрация устройств
![]() Получение и просмотр сертификатов от уполномоченных органов
Получение и просмотр сертификатов от уполномоченных органов
![]() Предоставить данные клинических исследований и исследований производительности
Предоставить данные клинических исследований и исследований производительности
![]() Сообщить о бдительности и послепродажном надзоре
Сообщить о бдительности и послепродажном надзоре
![]() Деятельность по надзору за рынком
Деятельность по надзору за рынком
Посмотрите комплексные вебинары по правилам медицинского оборудования
[Веб-трансляция]: Соответствие требованиям UDI для медицинских устройств GS1 и SoftGroup

Запросить веб-трансляцию