Dispositivos médicos
A rastreabilidade tem um papel fundamental no monitoramento, supervisão e melhoria da qualidade geral de dispositivos médicos e dispositivos in vitro.
Regulamentos da UE
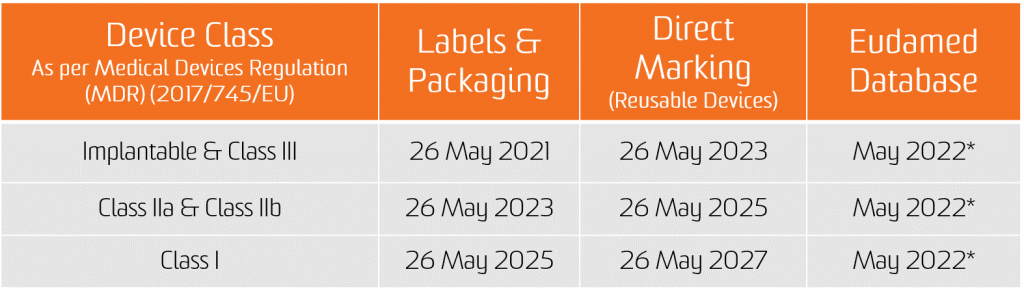
Em 2017 o Regulamento de Dispositivos Médicos (MDR) foi publicado e marcou o início de uma jornada de quatro anos de transição de MDD para AIMDD. A partir de 26 de maio de 2021, o MDR será totalmente aplicável. O MDR afeta as indústrias de fabricação, distribuição ou aquisição de dispositivos médicos.
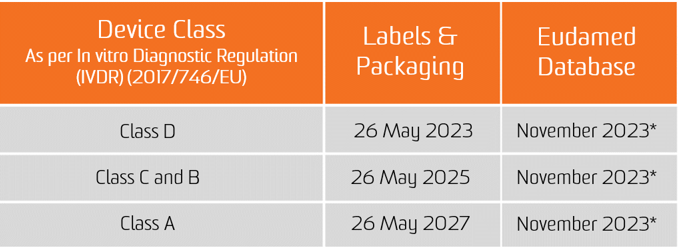
O Regulação de diagnóstico in vitro (IVDR) entrou em vigor a 25 de maio de 2017. A partir de 26 de maio de 2022, os novos dispositivos terão de cumprir os requisitos do IVDR para poderem ser colocados no mercado europeu. Os produtos já certificados por um Organismo Notificado podem ser colocados no mercado até 25 de maio de 2024 sob algumas condições e se o fabricante cumprir os pré-requisitos específicos traçados no IVDR.
O que é UDI?
UDI (Exclusivo Identificação do dispositivo)
O UDI é uma série de caracteres numéricos ou alfanuméricos que são criados por meio de um padrão de codificação e identificação de dispositivo aceito globalmente. Permite a identificação inequívoca de um dispositivo específico no mercado. A UDI é composta pela UDI-DI e pela UDI-PI.
 UDI-DI (GTIN Equivalente GS1 - Número Global de Item Comercial)
UDI-DI (GTIN Equivalente GS1 - Número Global de Item Comercial)
O UDI-DI é um código numérico ou alfanumérico único específico para um modelo do dispositivo e que também é usado como a 'chave de acesso' às informações armazenadas em um banco de dados UDI.
UDI-PI (IA Equivalente GS1 - Identificadores de Aplicação)
O UDI-PI é um código numérico ou alfanumérico que identifica a unidade de produção do dispositivo.
Os diferentes tipos de UDI-PIs incluem um número de série, número de lote, identificação de software e data de fabricação ou expiração, ou ambos os tipos de dados.
Implementação UDI
A obrigação de atribuição de UDI aplica-se a partir da data de aplicação dos dois novos Regulamentos, ou seja, 26 de maio de 2021 para os dispositivos médicos e 26 de maio de 2022 para os dispositivos médicos de diagnóstico in vitro.
A obrigação de submissão de dados UDI na base de dados da EUDAMED aplica-se a partir de 26 de novembro de 2022 para dispositivos médicos e 26 de novembro de 2023 para dispositivos médicos de diagnóstico in vitro (desde que a EUDAMED esteja em pleno funcionamento antes da data de aplicação do respetivo Regulamento; caso contrário, esta obrigação aplica-se 24 meses após a entrada em pleno funcionamento da EUDAMED).
No entanto, os fabricantes poderão cumprir voluntariamente as obrigações de registro a partir de 26 de maio de 2021 para dispositivos médicos e 26 de maio de 2022 para dispositivos médicos de diagnóstico in vitro.
Deve-se observar que, desde que o Eudamed esteja totalmente funcional, a qualquer momento após 26 de maio de 2021 para dispositivos médicos e 26 de maio de 2022 para dispositivos médicos de diagnóstico in vitro, o registro completo de dispositivos (artigo 29 do MDR e artigo 26 do IVDR) continua a ser uma pré-condição para o possível registro de seu incidente grave relevante na Eudamed.
*A edição do CAMD em 11 de novembro de 2020 afirma que a Comissão Europeia está anunciando o adiamento do lançamento do Eudamed como um todo até maio de 2022 por motivos legais. A nova data de lançamento do Eudamed coincide com a data de implementação do Regulamento de Diagnóstico In Vitro, previsto para entrar em vigor em 26 de maio de 2022.
O que é EUDAMED?
A nova base de dados europeia regulamentar sobre dispositivos médicos
Eudamed é um portal seguro baseado na web que atua como um repositório central para troca de informações entre as Autoridades Competentes nacionais e a Comissão, de acordo com os Regulamentos MDR e IVDR.
O principal papel da Eudamed inclui as seguintes responsabilidades:
![]() Registro Eudamed
Registro Eudamed
![]() Registro de UDI/Dispositivos
Registro de UDI/Dispositivos
![]() Obtenha e visualize certificados de órgãos notificados
Obtenha e visualize certificados de órgãos notificados
![]() Fornecer dados de investigações clínicas e estudos de desempenho
Fornecer dados de investigações clínicas e estudos de desempenho
![]() Vigilância de relatórios e vigilância pós-comercialização
Vigilância de relatórios e vigilância pós-comercialização
![]() Atividades de vigilância do mercado
Atividades de vigilância do mercado
Assista aos webinars completos para Regulamentos de Dispositivos Médicos
[Webcast]: Atendendo ao requisito de UDI para dispositivos médicos pela GS1 e SoftGroup

Solicite o webcast