欧盟口蹄疫概况
随着 2011 年 7 月发布的伪造药品指令 (FMD), 欧洲药品管理局 (EMA) 开始制定欧盟国家的追溯标准。
这 伪造药品指令(指令 2011/62/EU) 引入统一的欧洲措施来打击药品造假并确保药品安全并严格控制贸易流通中的药品。措施包括:
- 药品外包装上的强制性安全特征——唯一标识符和防篡改装置
- 用于识别合法在线药店的欧盟通用徽标
- 更严格的活性药物成分进口规定
- 加强对批发分销商的记录保存要求
截至 2019 年,所有医药产品都必须完全满足 FMD 义务。到 2025 年,希腊和意大利等拥有独立系统的欧盟国家必须完全遵守药品跟踪和追溯法规。
序列化要求
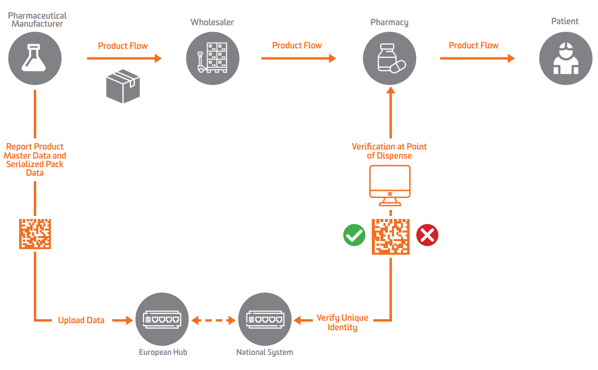
根据 EU FMD,序列化必须出现在欧洲的二级或可销售单位级别。为了实现验证,制造商需要首先对产品进行序列化,并将序列化的数据发送到可以对其执行查询的中央存储库。
为了实现序列化、验证并向当局报告,欧盟 FMD 要求制造商在包装上标记四个数据元素,这些数据元素必须以人类可读的形式打印,并编码并存储在 GS1 2D DataMatrix 中:
- 产品标识符
- 序列号
- 批号
- 到期日
第五个数据元素(国家报销编号)的使用是可选的,欧盟很少有国家可能要求在唯一标识符中包含相同的数据元素,以链接社会化医疗计划下药品的报销。
药品制造商和水货商必须 向中央欧盟中心报告数据 由 欧洲药品验证组织 (EMVO),它还将用户载入 Hub。这会将数据推送到相应的国家药品验证组织 (NMVO) 运行的适当数据存储库,这些组织负责最终用户的入职和国家系统的运行。
报告要求
根据 EU FMD,上市许可持有人 (MAH) 需要提交产品主数据和序列化产品包装数据。
主数据包括:
- 产品代码
- 形式
- 力量
- 每包剂量
- 包装类型
- 分销目标市场
- 产品主数据的未来和任何更新
序列化产品包数据包括:
- 产品代码
- 批号/批号
- 到期日
- 序列号
- 序列化产品包数据的任何更新