Общ преглед на шапа в ЕС
С Директивата за фалшифицираните лекарства (FMD), публикувана през юли 2011 г., Европейска агенция по лекарствата (EMA) поставя началото на стандартите за проследимост на страните в Европейския съюз.
The Директива относно фалшифицираните лекарства (Директива 2011/62/ЕС) въвежда хармонизирани европейски мерки за борба с фалшифицирането на лекарства и гарантира, че лекарствата са безопасни и че лекарствата в търговско обращение са строго контролирани. Мерките включват:
- Задължителни характеристики за безопасност – уникален идентификатор и устройство против подправяне – върху външната опаковка на лекарствата
- Общо лого за целия ЕС за идентифициране на законни онлайн аптеки
- По-строги правила за внос на активни фармацевтични съставки
- Засилени изисквания за водене на документация за дистрибуторите на едро
От 2019 г. всички фармацевтични продукти трябва да отговарят напълно на задълженията за шап. До 2025 г. страните от ЕС с отделна система, като Гърция и Италия, трябва да бъдат напълно съвместими с регламента за проследяване и проследяване на фармацевтични продукти.
Изисквания за сериализация
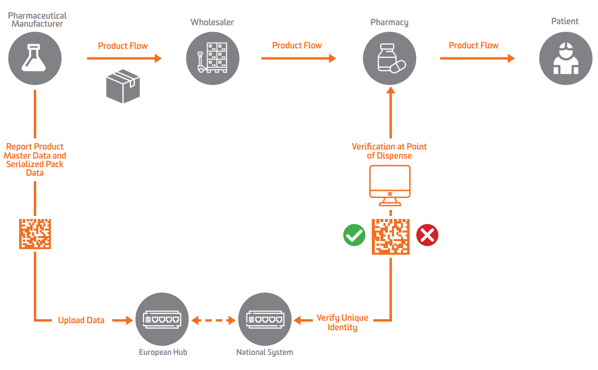
Според EU FMD, сериализирането трябва да се появи на ниво вторично или продаваемо устройство в Европа. За да активират проверката, производителите трябва първо да сериализират продукта и да изпратят тези сериализирани данни до централно хранилище, което може да извършва заявки срещу него.
За да се даде възможност за сериализиране, проверка и докладване на властите, EU FMD изисква производителите да маркират пакетите с четири елемента от данни, които трябва да бъдат отпечатани в четима от хора форма и кодирани и съхранени в GS1 2D DataMatrix:
- Идентификатор на продукта
- Сериен номер
- Номер на партида или партида
- Срок на годност
Използването на петия елемент от данни — националния номер за реимбурсиране — не е задължително и малко държави в ЕС могат да поискат включване на същия в уникален идентификатор, за да се свърже реимбурсирането на лекарствен продукт по програма за социализирана медицина.
Фармацевтичните производители и паралелните търговци трябва докладвайте данни на централен център на ЕС управляван от Европейска организация за проверка на лекарствата (EMVO), които също включват потребители в Hub. Това ще насочи данните към подходящи хранилища на данни, управлявани от съответните национални организации за проверка на лекарствата (NMVO), които отговарят за включването на крайния потребител и за работата на националните системи.
Изисквания за докладване
Съгласно шапа в ЕС притежателят на разрешението за употреба (ПРУ) е длъжен да предостави основни данни за продукта и сериализирани данни за опаковката на продукта.
Основните данни включват:
- Продуктови кодове
- форма
- Сила
- Дози в опаковка
- Тип опаковка
- Целеви пазар(и) за разпространение
- Бъдещи и всякакви актуализации на основни данни за продукта
Сериализираните данни за продуктовия пакет включват:
- Продуктови кодове
- Партиден номер
- Срок на годност
- Серийни номера
- Всякакви актуализации на сериализирани данни за продуктов пакет