Visão geral da febre aftosa da UE
Com a Diretiva de Medicamentos Falsificados (FMD) publicada em julho de 2011, o Agência Europeia de Medicamentos (EMA) marcaram o início dos padrões de rastreabilidade dos países da União Européia.
O Diretiva de Medicamentos Falsificados (Diretiva 2011/62/UE) introduz medidas europeias harmonizadas para combater a falsificação de medicamentos e garantir que os medicamentos sejam seguros e que os medicamentos em circulação sejam rigorosamente controlados. As medidas incluem:
- Dispositivos de segurança obrigatórios – um identificador único e um dispositivo anti-adulteração – na embalagem exterior dos medicamentos
- Um logotipo comum em toda a UE para identificar farmácias on-line legais
- Regras mais rígidas para importação de insumos farmacêuticos ativos
- Requisitos de manutenção de registros reforçados para distribuidores atacadistas
A partir de 2019, todos os produtos farmacêuticos devem cumprir integralmente as obrigações da febre aftosa. Até 2025, os países da UE com um sistema separado, como a Grécia e a Itália, devem estar em total conformidade com o regulamento de rastreamento e rastreamento farmacêutico.
Requisitos de serialização
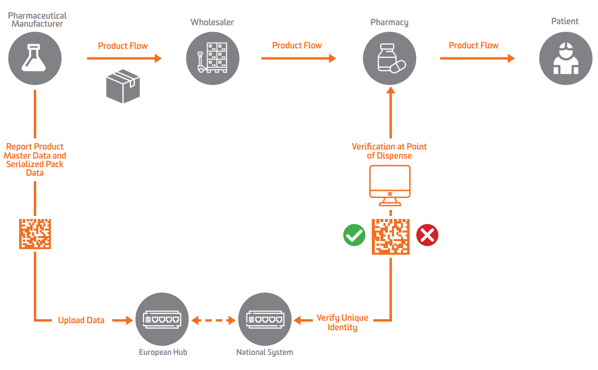
De acordo com a EU FMD, a serialização deve aparecer no nível secundário ou de unidade vendável na Europa. Para habilitar a verificação, os fabricantes precisam primeiro serializar o produto e enviar esses dados serializados para um repositório central que pode realizar consultas nele.
Para permitir a serialização, verificação e comunicação às autoridades, a FMD da UE exige que os fabricantes marquem as embalagens com quatro elementos de dados que devem ser impressos em formato legível por humanos e codificados e armazenados numa DataMatrix 2D GS1:
- identificador de produto
- Número de série
- Lote ou número do lote
- Data de validade
A utilização do quinto elemento de dados — o número de reembolso nacional — é opcional e poucos estados da UE podem solicitar a inclusão do mesmo num identificador único para associar o reembolso de um medicamento ao abrigo de um programa de medicina socializada.
Os fabricantes farmacêuticos e os comerciantes paralelos devem relatar dados para um hub central da UE executado pelo Organização Europeia de Verificação de Medicamentos (EMVO), que também integra usuários ao Hub. Isso enviará os dados para repositórios de dados apropriados administrados pelas Organizações Nacionais de Verificação de Medicamentos (NMVOs) correspondentes, que são responsáveis pela integração do usuário final e pela operação dos sistemas nacionais.
Requisitos de relatórios
De acordo com a FMD da UE, o Titular da Autorização de Introdução no Mercado (MAH) é obrigado a enviar os dados mestre do produto e os dados serializados da embalagem do produto.
Os dados mestre incluem:
- códigos de produto
- Forma
- Força
- Doses por embalagem
- tipo de embalagem
- Mercado(s)-alvo para distribuição
- Futuro e quaisquer atualizações nos dados mestre do produto
Os dados serializados do pacote de produtos incluem:
- códigos de produto
- Número do lote
- Data de validade
- números de série
- Quaisquer atualizações nos dados do pacote de produtos serializados
Descubra como o SoftGroup pode ajudá-lo a cumprir a conformidade com a febre aftosa da UE
Ver soluções